Plasmodium P36
Plasmodium P36 Modulates Host Cell Receptor Selection During Sporozoite Invasion
Plasmodium P36 Modulates Host Cell Receptor Selection During Sporozoite Invasion
Plasmodium P36 has an important role in malaria infection, specifically in the initial stages of host cell invasion by sporozoites. Recent studies have shown that P36 interacts with host cell receptors, influencing the parasite's ability to invade and establish infection. By modulating host cell receptor selection, P36 plays a role in determining the efficiency and success of sporozoite invasion.
The molecular mechanisms behind P36-mediated invasion is important for the parasite's life cycle.
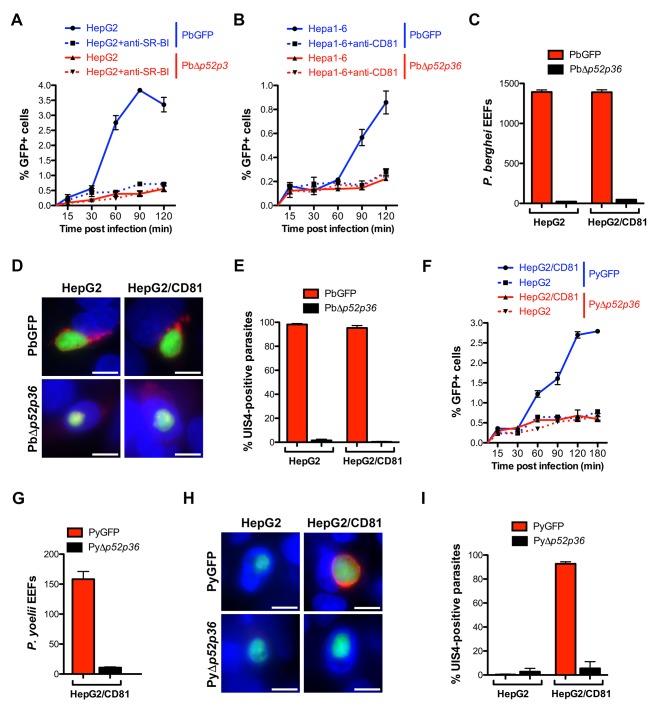
Sporozoite productive invasion irrespective of the host cell entry pathway:
The 6-cysteine domain proteins P52 and P36 are indispensable for the productive invasion of sporozoites, regardless of the host cell entry pathway. These proteins are situated on the surface of the sporozoites and play a crucial role in recognizing and binding to host cell receptors, facilitating invasion. Their absence severely impairs the ability of sporozoites to invade host cells efficiently. Studies have demonstrated that P52 and P36 interact with various components of the invasion machinery, highlighting their involvement in multiple stages of the invasion process. This intricate interplay underscores the significance of 6-cysteine domain proteins in mediating sporozoite invasion, emphasizing their potential as therapeutic targets for combating parasitic infections.